![]()
MnM, a machine learning approach to detect replication states and genomic subpopulations for single-cell DNA replication timing disentanglement from whole-genome sequencing data. It includes a single-cell copy-number imputation method, a replication state classifier and a subpopulation detector. Requires ≥10 cells.
# Move to the downloaded directory
cd path_to_directory
# Make MnM executable
chmod +x MnM
# See all options and explanations
./MnM -h
# Launch
./MnM -i INPUTFILE [-m] [--sep SEP] [-o OUTPUT] [-n NAME] [-g GENOME] [-w WINDOWSIZE] [--seed SEED] [--maxcells MAXCELLS] [-r] [-s] [--cpu CPU] [--CNcol CNCOL] [--Cellcol CELLCOL] [--groups GROUPS] [-p] [-b] [-v] [-h]Processed CNV data of MCF-7 cells obtained from published data with Kronos scRT can be used as an example. These data are included in the scRT/scCNV atlas. We can discover replication states and genomic heterogeneity with the following command:
MnM -i scRT_scCNV_Atlas/Gnan2022_MCF-7/Gnan2022_MCF-7_scCNV_Matrix.tsv.gz -o ~/Documents/MnM_test_MCF-7_MnM_Output -m -g hg38 -n MCF-7 -r -sWhere -i is the input file (scCNV matrix), -o the output directory, -m flags that the input file is a matrix and not a BAM file, -g precises the reference genome, -n names the output files, -r reports replication states and -s discovers genomic heterogeneity (subpopulations).
As a result the following figures are produced:
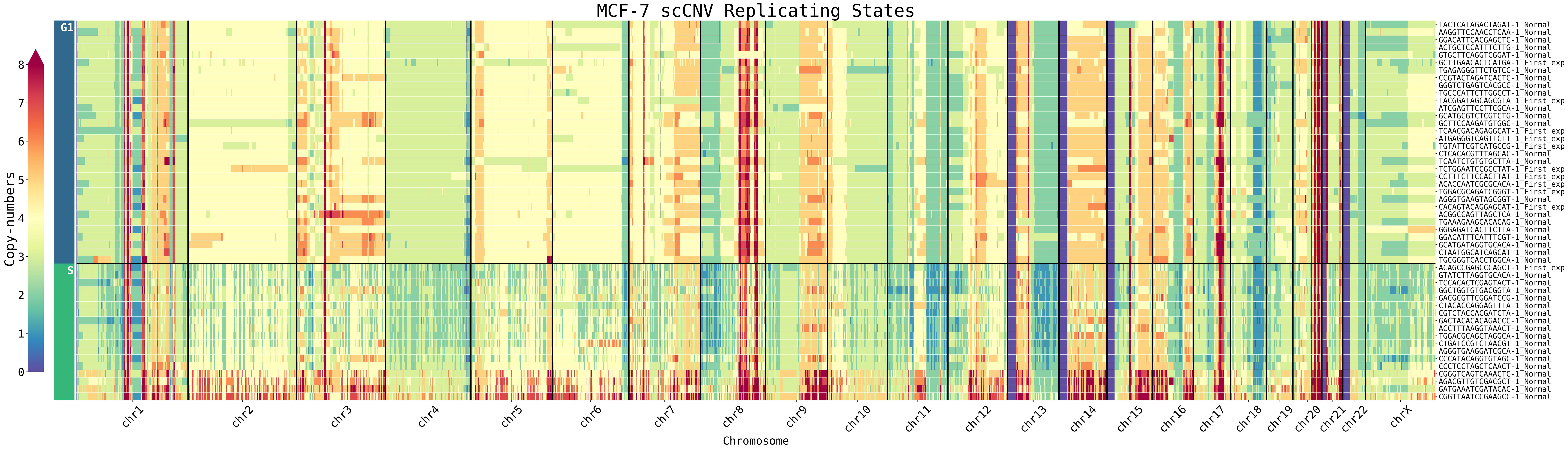
- Distinction of replicating cells from non-replicating cells visualised on a genome-wide single-cell plot.
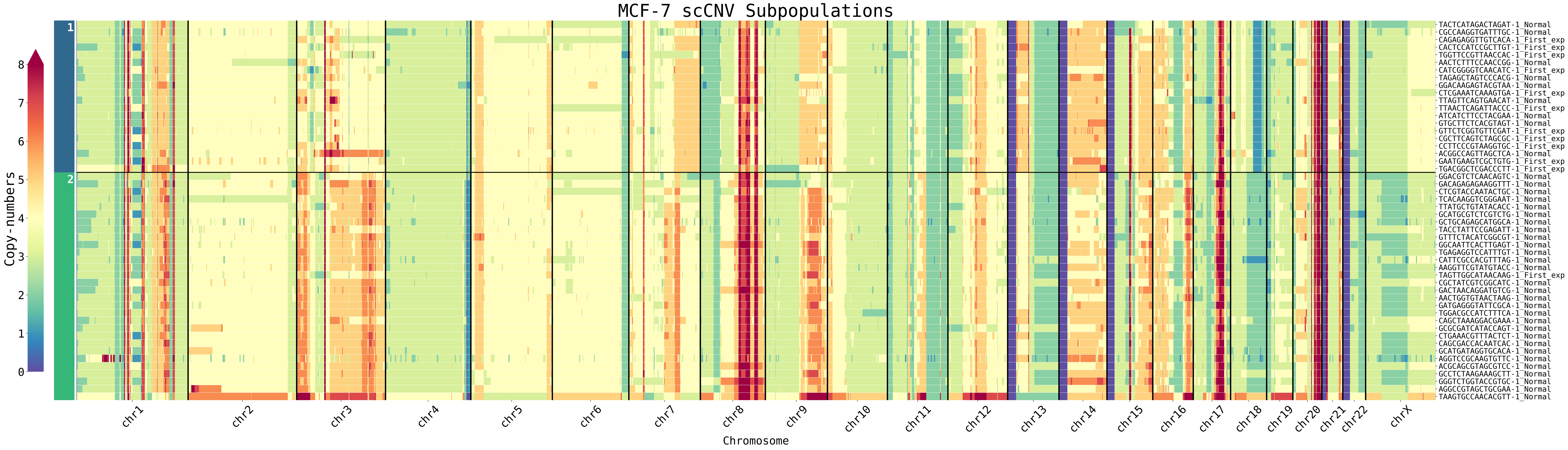
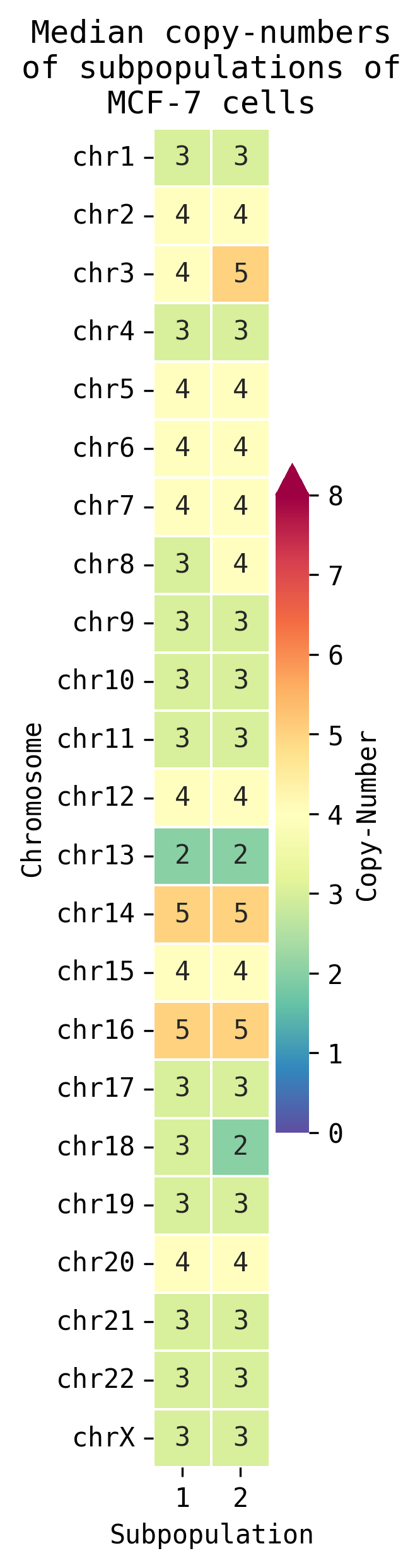
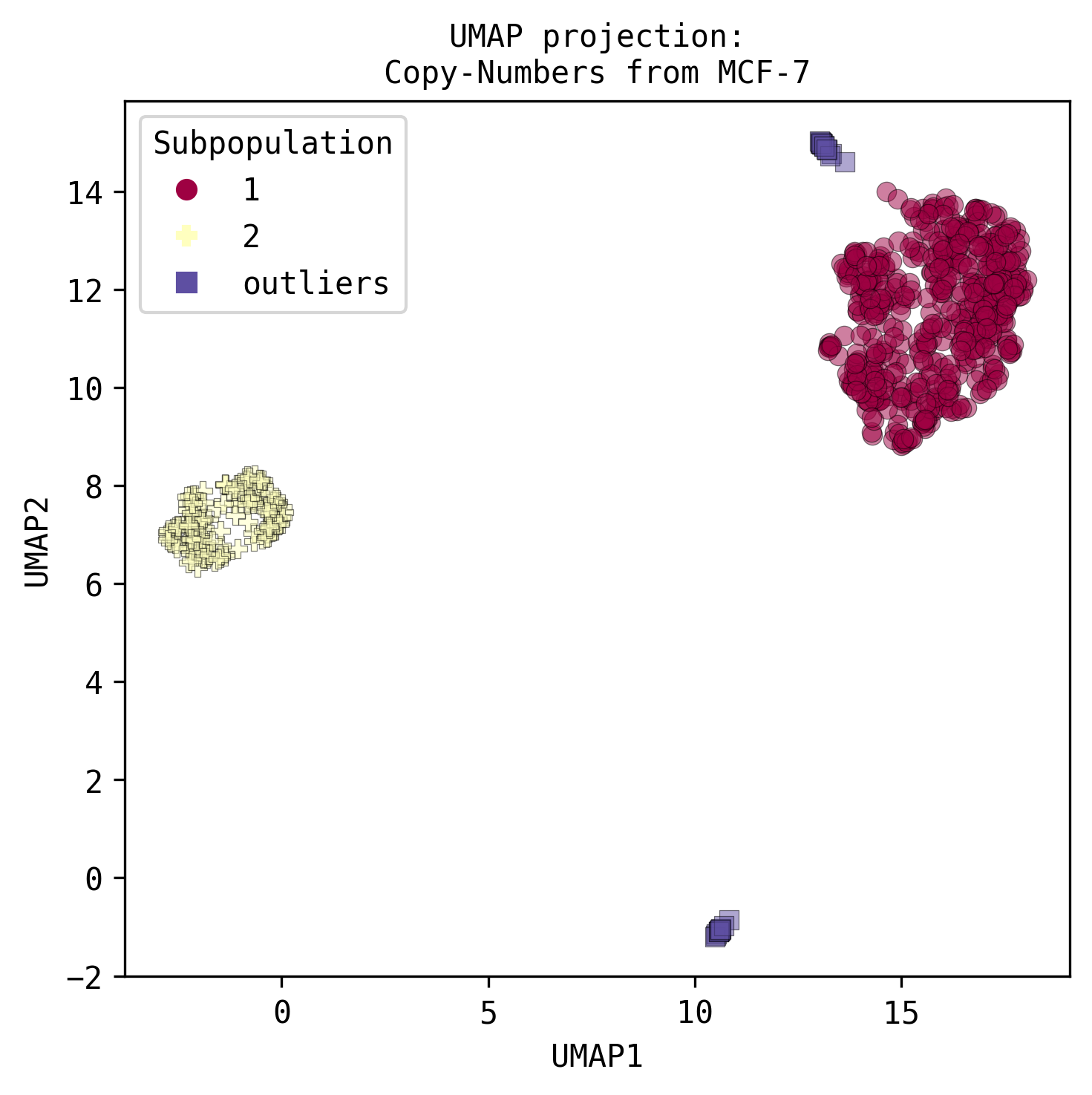
- Detection of subpopulations from non-replicating cells visualised:
- Genome-wide at the single-cell level.
- Chromosome-wide per subpopulation.
- On a UMAP plot.
And a metadata file lists the replication state and subpopulation of each cell:
| Cell | Phase | Subpopulation |
|---|---|---|
| AAACCTGCAACCCAAT-1_First_exp.bam | G1 | 1 |
| AAACCTGGTCATTACG-1_Normal.bam | S | 1 |
| AAACGGGTCGGGAAAC-1_Normal.bam | G1 | 2 |
| AAAGATGAGCTATGCT-1_Normal.bam | S | 1 |
| AAAGATGAGTAAGTAC-1_Normal.bam | S | 2 |
| AAAGATGGTTTGCCTC-1_Normal.bam | G1 | 1 |
| CTAGTGAAGTGCTGCC-1_Normal.bam | S | 1 |
| ... | ... | ... |
This software is provided as-is and without warranty. By using this software, you agree to abide by the terms specified in the license.
This program has been tested with the following software versions. It is recommended you use the same or superior versions.
- Python 3.9.11
- pandas 1.5.0
- numpy 1.24.3
- sklearn 1.1.2
- natsort 8.2.0
- seaborn 0.12.1
- matplotlib 3.6.2
- pybedtools 0.9.0
- multiprocess 0.70.14
- tensorflow 2.13.0
- umap 0.5.3
You can run:
pip install pandas==1.5.0
pip install numpy==1.24.3
pip install scikit-learn==1.1.2
pip install natsort==8.2.0
pip install seaborn==0.12.1
pip install matplotlib==3.6.2
pip install pybedtools==0.9.0
pip install multiprocess==0.70.14
pip install tensorflow==2.13.0
pip install umap==0.5.3If you use MnM please cite our publication:
Josephides, J.M., Chen, CL. Unravelling single-cell DNA replication timing dynamics using machine learning reveals heterogeneity in cancer progression. Nat Commun 16, 1472 (2025). https://doi.org/10.1038/s41467-025-56783-0.
For inquiries and authorization requests, please contact chunlong.chen@curie.fr.
MnM v1.0.1
Author: Joseph Josephides. Institut Curie, Paris.